I этап – подготовка документов
Определение класса риска и номенклатурного вида
- класс риска применения МИ – см. Решение №173 «Об утверждении Правил классификации медицинских изделий в зависимости от потенциального риска применения».
- номенклатурный вид МИ (см. https://portal.eaeunion.org/sites/odata/redesign/Pages/MedicalDeviceNomenclature.aspx)
Подготовка документов
зависит от класса потенциального риска применения МИ, см в Решении №144 «О внесении изменений в Правила регистрации и экспертизы безопасности, качества и эффективности МИ»
-
Класс потенциального риска: 1 — Низкая степень
|
№ п/п |
Наименование документа |
Условия предоставления |
Примечание |
Форма документа |
|
1 |
Заявление по формам, предусмотренным приложениями № 2 и 3 к Правилам регистрации и экспертизы безопасности, качества и эффективности медицинских изделий ( №46 или 144) |
Обязательно |
Решение 144, приложение 2 |
|
|
2 |
Доверенность от производителя на право представления интересов при регистрации |
При необходимости |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства – члена Евразийского экономического союза (далее – государство-член) |
|
|
3 |
Копия разрешительного документа на право производства в стране-производителе с приложением |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
4 |
Копии сертификатов на систему менеджмента качества производителя медицинских изделий (ИСО 13485 либо соответствующий региональный или национальный стандарт государства-члена) |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
5 |
Декларация о соответствии требованиям безопасности и эффективности медицинских изделий или эквивалентный документ |
При наличии |
||
|
6 |
Копия регистрационного удостоверения (сертификата свободной продажи, сертификата на экспорт (за исключением медицинских изделий, впервые произведенных на территории государства-члена)), выданного в стране производителя (при наличии) с представлением перевода на русский язык |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
7 |
Копия документа, удостоверяющего регистрацию в других странах |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
8 |
Справка на медицинское изделие с описанием области применения, назначения, краткой характеристики медицинского изделия, вариантами исполнения и комплектующими (по форме) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
9 |
Данные о маркировке и упаковке (полноцветные макеты упаковок и этикеток, текст маркировки на русском языке и государственных языках государств-членов) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
10 |
Информация о разработке и производстве: схемы процессов производства, основные стадии производства, упаковка, испытания и процедура выпуска конечного продукта |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
11 |
Сведения о производителе: наименование, вид деятельности, юридический адрес, форма собственности, состав руководства, перечень подразделений и дочерних компаний с указанием их статуса и полномочий |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
12 |
Сообщения о несчастных случаях и отзывах (информация не предоставляется для вновь разработанных и спроектированных медицинских изделий): список нежелательных событий или несчастных случаев, связанных с использованием изделия, и указание периода времени, на протяжении которого происходили указанные случаи если нежелательных событий слишком много, необходимо предоставить краткие обзоры по каждому из типов событий и указать общее количество событий каждого типа, о которых поступали отчеты список отзывов с рынка медицинских изделий и (или) пояснительных уведомлений и описание подхода к рассмотрению этих проблем и их решению производителями в каждом из таких случаев описание анализа и (или) корректирующих действий, предпринятых в ответ на указанные случаи |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
13 |
Перечень стандартов, которым соответствует медицинское изделие (с указанием сведений о них) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
14 |
Сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них (далее – общие требования) |
Обязательно |
||
|
15 |
Документ, устанавливающий требования к техническим характеристикам медицинского изделия |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
16 |
Протоколы технических испытаний, проведенных в целях доказательства соответствия общим требованиям |
Обязательно |
||
|
17 |
Протоколы исследований (испытаний) по оценке биологического действия медицинского изделия, проведенных в целях доказательства соответствия общим требованиям |
Обязательно |
||
|
18 |
Отчет о клиническом доказательстве эффективности и безопасности медицинского изделия |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
19 |
Отчет об анализе рисков |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
20 |
Данные о лекарственных средствах в составе медицинского изделия (состав лекарственного средства, количество, данные о совместимости лекарственного средства с медицинским изделием, регистрации лекарственного средства в стране-производителе) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
21 |
Данные о биологической безопасности |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
22 |
Данные о процедуре стерилизации, включая информацию о валидации процесса, результаты тестирования на содержание микроорганизмов (степень биологической нагрузки), пирогенности, стерильности (при необходимости) с указанием методов проведения испытаний и данные о валидации упаковки (для стерильных изделий) |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
23 |
Информация о специальном программном обеспечении (при наличии): сведения производителя о валидации программного обеспечения |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
24 |
Отчет об исследованиях стабильности – с аутентичным переводом на русский язык результатов и выводов испытаний для изделий, имеющих срок хранения |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
25 |
Эксплуатационный документ или инструкция по применению медицинского изделия на государственном языке государств признания (при необходимости) и русском языках |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
26 |
Руководство по сервисному обслуживанию (в части комплектующих медицинского изделия) – в случае отсутствия данных в эксплуатационной документации |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
27 |
Отчет об инспекции производства |
При наличии |
||
|
28 |
План сбора и анализа данных по безопасности и эффективности медицинских изделий на постпродажном этапе |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
-
Класс потенциального риска: 2а — Средняя степень
|
№ п/п |
Наименование документа |
Условия предоставления |
Примечание |
Форма документа |
|---|---|---|---|---|
|
1 |
Заявление по формам, предусмотренным приложениями № 2 и 3 к Правилам регистрации и экспертизы безопасности, качества и эффективности медицинских изделий |
Обязательно |
||
|
2 |
Доверенность от производителя на право представления интересов при регистрации |
При необходимости |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства – члена Евразийского экономического союза (далее – государство-член) |
|
|
3 |
Копия разрешительного документа на право производства в стране-производителе с приложением |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
4 |
Копии сертификатов на систему менеджмента качества производителя медицинских изделий (ИСО 13485 либо соответствующий региональный или национальный стандарт государства-члена) |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
5 |
Декларация о соответствии требованиям безопасности и эффективности медицинских изделий или эквивалентный документ |
При наличии |
||
|
6 |
Копия регистрационного удостоверения (сертификата свободной продажи, сертификата на экспорт (за исключением медицинских изделий, впервые произведенных на территории государства-члена)), выданного в стране производителя (при наличии) с представлением перевода на русский язык |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
7 |
Копия документа, удостоверяющего регистрацию в других странах |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
8 |
Справка на медицинское изделие с описанием области применения, назначения, краткой характеристики медицинского изделия, вариантами исполнения и комплектующими (по форме) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
9 |
Данные о маркировке и упаковке (полноцветные макеты упаковок и этикеток, текст маркировки на русском языке и государственных языках государств-членов) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
10 |
Информация о разработке и производстве: схемы процессов производства, основные стадии производства, упаковка, испытания и процедура выпуска конечного продукта |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
11 |
Сведения о производителе: наименование, вид деятельности, юридический адрес, форма собственности, состав руководства, перечень подразделений и дочерних компаний с указанием их статуса и полномочий |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
12 |
Сообщения о несчастных случаях и отзывах (информация не предоставляется для вновь разработанных и спроектированных медицинских изделий): список нежелательных событий или несчастных случаев, связанных с использованием изделия, и указание периода времени, на протяжении которого происходили указанные случаи если нежелательных событий слишком много, необходимо предоставить краткие обзоры по каждому из типов событий и указать общее количество событий каждого типа, о которых поступали отчеты список отзывов с рынка медицинских изделий и (или) пояснительных уведомлений и описание подхода к рассмотрению этих проблем и их решению производителями в каждом из таких случаев описание анализа и (или) корректирующих действий, предпринятых в ответ на указанные случаи |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
13 |
Перечень стандартов, которым соответствует медицинское изделие (с указанием сведений о них) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
14 |
Сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них (далее – общие требования) |
Обязательно |
||
|
15 |
Документ, устанавливающий требования к техническим характеристикам медицинского изделия |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
16 |
Протоколы технических испытаний, проведенных в целях доказательства соответствия общим требованиям |
Обязательно |
||
|
17 |
Протоколы исследований (испытаний) по оценке биологического действия медицинского изделия, проведенных в целях доказательства соответствия общим требованиям |
Обязательно |
||
|
18 |
Отчет о клиническом доказательстве эффективности и безопасности медицинского изделия |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
19 |
Отчет об анализе рисков |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
20 |
Данные о лекарственных средствах в составе медицинского изделия (состав лекарственного средства, количество, данные о совместимости лекарственного средства с медицинским изделием, регистрации лекарственного средства в стране-производителе) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
21 |
Данные о биологической безопасности |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
22 |
Данные о процедуре стерилизации, включая информацию о валидации процесса, результаты тестирования на содержание микроорганизмов (степень биологической нагрузки), пирогенности, стерильности (при необходимости) с указанием методов проведения испытаний и данные о валидации упаковки (для стерильных изделий) |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
23 |
Информация о специальном программном обеспечении (при наличии): сведения производителя о валидации программного обеспечения |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
24 |
Отчет об исследованиях стабильности – с аутентичным переводом на русский язык результатов и выводов испытаний для изделий, имеющих срок хранения |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
25 |
Эксплуатационный документ или инструкция по применению медицинского изделия на государственном языке государств признания (при необходимости) и русском языках |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
26 |
Руководство по сервисному обслуживанию (в части комплектующих медицинского изделия) – в случае отсутствия данных в эксплуатационной документации |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
27 |
Отчет об инспекции производства |
При наличии |
||
|
28 |
План сбора и анализа данных по безопасности и эффективности медицинских изделий на постпродажном этапе |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
-
Класс потенциального риска: 3 — Высокая степень
|
№ п/п |
Наименование документа |
Условия предоставления |
Примечание |
Форма документа |
|---|---|---|---|---|
|
1 |
Заявление по формам, предусмотренным приложениями № 2 и 3 к Правилам регистрации и экспертизы безопасности, качества и эффективности медицинских изделий |
Обязательно |
||
|
2 |
Доверенность от производителя на право представления интересов при регистрации |
При необходимости |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства – члена Евразийского экономического союза (далее – государство-член) |
|
|
3 |
Копия разрешительного документа на право производства в стране-производителе с приложением |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
4 |
Копии сертификатов на систему менеджмента качества производителя медицинских изделий (ИСО 13485 либо соответствующий региональный или национальный стандарт государства-члена) |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
5 |
Декларация о соответствии требованиям безопасности и эффективности медицинских изделий или эквивалентный документ |
При наличии |
||
|
6 |
Копия регистрационного удостоверения (сертификата свободной продажи, сертификата на экспорт (за исключением медицинских изделий, впервые произведенных на территории государства-члена)), выданного в стране производителя (при наличии) с представлением перевода на русский язык |
При наличии |
Документ предоставляется в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена |
|
|
7 |
Копия документа, удостоверяющего регистрацию в других странах |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
8 |
Справка на медицинское изделие с описанием области применения, назначения, краткой характеристики медицинского изделия, вариантами исполнения и комплектующими (по форме) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
9 |
Данные о маркировке и упаковке (полноцветные макеты упаковок и этикеток, текст маркировки на русском языке и государственных языках государств-членов) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
10 |
Информация о разработке и производстве: схемы процессов производства, основные стадии производства, упаковка, испытания и процедура выпуска конечного продукта |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
11 |
Сведения о производителе: наименование, вид деятельности, юридический адрес, форма собственности, состав руководства, перечень подразделений и дочерних компаний с указанием их статуса и полномочий |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
12 |
Информация о маркетинге (история при условии обращения изделия на рынке более 2 лет) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
13 |
Сообщения о несчастных случаях и отзывах (информация не предоставляется для вновь разработанных и спроектированных медицинских изделий): список нежелательных событий или несчастных случаев, связанных с использованием изделия, и указание периода времени, на протяжении которого происходили указанные случаи если нежелательных событий слишком много, необходимо предоставить краткие обзоры по каждому из типов событий и указать общее количество событий каждого типа, о которых поступали отчеты список отзывов с рынка медицинских изделий и (или) пояснительных уведомлений и описание подхода к рассмотрению этих проблем и их решению производителями в каждом из таких случаев описание анализа и (или) корректирующих действий, предпринятых в ответ на указанные случаи |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
14 |
Перечень стандартов, которым соответствует медицинское изделие (с указанием сведений о них) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
15 |
Сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них (далее – общие требования) |
Обязательно |
||
|
16 |
Документ, устанавливающий требования к техническим характеристикам медицинского изделия |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
17 |
Протоколы технических испытаний, проведенных в целях доказательства соответствия общим требованиям |
Обязательно |
||
|
18 |
Протоколы исследований (испытаний) по оценке биологического действия медицинского изделия, проведенных в целях доказательства соответствия общим требованиям |
Обязательно |
||
|
19 |
Отчет о клиническом доказательстве эффективности и безопасности медицинского изделия |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
20 |
Отчет об анализе рисков |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
21 |
Данные о лекарственных средствах в составе медицинского изделия (состав лекарственного средства, количество, данные о совместимости лекарственного средства с медицинским изделием, регистрации лекарственного средства в стране-производителе) |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
22 |
Данные о биологической безопасности |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
23 |
Данные о процедуре стерилизации, включая информацию о валидации процесса, результаты тестирования на содержание микроорганизмов (степень биологической нагрузки), пирогенности, стерильности (при необходимости) с указанием методов проведения испытаний и данные о валидации упаковки (для стерильных изделий) |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
24 |
Информация о специальном программном обеспечении (при наличии): сведения производителя о валидации программного обеспечения |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
25 |
Отчет об исследованиях стабильности – с аутентичным переводом на русский язык результатов и выводов испытаний для изделий, имеющих срок хранения |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
26 |
Эксплуатационный документ или инструкция по применению медицинского изделия на государственном языке государств признания (при необходимости) и русском языках |
При необходимости |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
27 |
Руководство по сервисному обслуживанию (в части комплектующих медицинского изделия) – в случае отсутствия данных в эксплуатационной документации |
При наличии |
Документ заверяется производителем (его уполномоченным представителем) |
|
|
28 |
Отчет об инспекции производства |
При наличии |
||
|
29 |
План сбора и анализа данных по безопасности и эффективности медицинских изделий на постпродажном этапе |
Обязательно |
Документ заверяется производителем (его уполномоченным представителем) |
|
Сбор доказательств
для подтверждения соответствия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации (Решение №27 Об утверждении Общих требований безопасности и эффективности медицинских изделий, требований к их маркировке и эксплуатационной документации на них), заявитель проводит следующие виды испытаний:
- технические испытания (Решение 28 Об утверждении Правил проведения технических испытаний медицинских изделий)
- испытания (исследования) с целью оценки биологического действия медицинского изделия (Решение Совета ЕЭК № 38 Об утверждении Правил проведения исследований (испытаний) с целью оценки биологического действия медицинских изделий)
Испытания с целью оценки биологического действия медицинского изделия проводятся в отношении медицинских изделий и (или) принадлежностей к медицинским изделиям, контактирующих с поверхностью тела человека, его слизистыми оболочками, внутренними средами организма
- испытания в целях утверждения типа средств измерений (Порядком утверждения типа средств измерений см. Решение Совета ЕЭК № 42 Об утверждении перечня видов медицинских изделий, подлежащих отнесению при их регистрации к средствам измерений)
Испытания проводятся в отношении медицинских изделий, отнесенных к средствам измерений, перечень которых утверждается Комиссией
- клинические испытания (исследования) (Решение Совета ЕЭК № 29 О Правилах проведения клинических и клинико-лабораторных испытаний (исследований) медицинских изделий)
Указанные виды испытаний должны проводиться в выбранных заявителем учреждениях и организациях, включенных в единый реестр уполномоченных организаций, имеющих право проводить исследования (испытания) медицинских изделий в целях их регистрации (Единый реестр уполномоченных организаций Евразийского экономического союза, осуществляющих проведение исследований (испытаний) медицинских изделий в целях их регистрации (eaeunion.org))
Объем исследований (испытаний) медицинских изделий в целях их регистрации зависит от вида медицинских изделий: неактивные, активные, активные диагностические, имплантируемые медицинские изделия, медицинских изделий для диагностики in vitro.
При проведении исследований (испытаний) медицинских изделий в целях их регистрации могут использоваться стандарты, включенные в перечень стандартов, в результате применения которых на добровольной основе полностью или частично обеспечивается соблюдение соответствия медицинского изделия общим требованиям, а также техническая документация производителя медицинского изделия
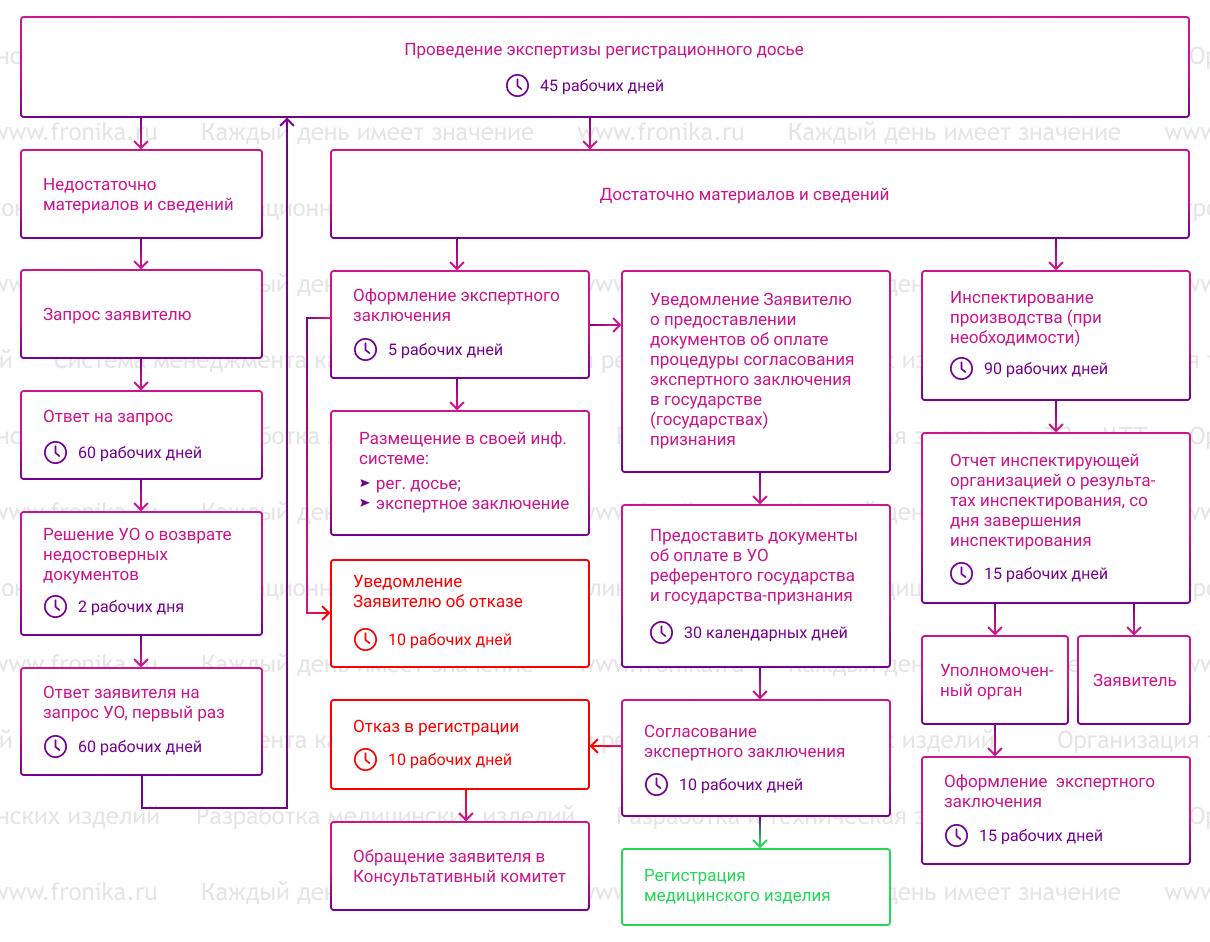
Схема проведения экспертизы регистрационного досьевключенных в перечень стандартов, с целью проведения технических испытаний медицинских изделий могут применяться методы (методики) испытаний, аттестованные (валидированные) и утвержденные в соответствии с законодательством государств - членов Союза
Уполномоченный орган (экспертная организация) или организация, определенная уполномоченным органом (экспертной организацией) референтного государства, проводит инспекцию производства медицинских изделий в соответствии с требованиями, установленными Комиссией.
Уполномоченный орган - орган государственной власти государства-члена, уполномоченный на осуществление и (или) координацию деятельности в сфере обращения медицинских изделий на территории этого государства-члена;
Инспекция производства медицинских изделий проводится до подготовки экспертного заключения
Выбор референтного государства и государства признания
Референтное государство - выбранное заявителем государство-член Союза, уполномоченный орган которого осуществляет регистрацию МИ
и, при необходимости регистрации медицинского изделия в нескольких государствах,
Государство признания - государство-член Союза, уполномоченный орган (экспертная организация) которого осуществляет процедуру согласования экспертного заключения референтного государства. Минимум одно государство-признания, см. решение 144, п.21.
Уполномоченные органы, предоставляющие услугу
Россия:
- Министерство здравоохранения Российской федерации;
- Федеральная служба по надзору в сфере здравоохранения.
Армения:
- Министерство здравоохранения Республики Армения;
- Научный центр экспертизы лекарств и медицинских технологий.
Беларусь:
- Министерство здравоохранения Республики Беларусь;
- Центр экспертиз и испытаний в здравоохранении Министерства здравоохранения Республики Беларусь.
Казахстан:
- Министерство здравоохранения и социального развития Республики Казахстан;
- Национальный центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники.
Киргизия:
- Министерство здравоохранения Кыргызской Республики.
Оплата госпошлины
за экспертизу и регистрацию медицинского изделия в референтном государстве
Оплата пошлин за экспертизу и регистрацию медицинского изделия в государствах признания производится после принятия положительного решения о регистрации медицинского изделия референтным государством.
Сайты государств-членов Союза, на которых возможно рассчитать размер пошлин и произвести их оплату:
- Россия: Министерство здравоохранения Российской федерации, Росздавназор;
- Армения: Министерство здравоохранения Республики Армения;
- Беларусь: Министерство здравоохранения Республики Беларусь;
- Казахстан: Министерство здравоохранения и социального развития Республики Казахстан;
- Киргизия: Министерство здравоохранения Кыргызской Республики.
II этап – регистрация и экспертиза МИ
Представление в УО документов
Заявитель представляет в уполномоченный орган УО (экспертную организацию) референтного государства следующие документы:
• заявление на проведение экспертизы и регистрации медицинского изделия (далее - заявление);
• документы по перечню согласно приложению N 4 , см. Решение 144;
• копии документов, подтверждающих оплату экспертизы и регистрации медицинского изделия в референтном государстве
7 рабочих дней – УО проверяет
полноту и достоверность предоставленных документов (реш.144, п.23), отправляет запрос заявителю о необходимости устранения выявленных нарушений (реш.144, п.24)
30 рабочих дней – дается заявителю на устранение
выявленных нарушений и (или) представления отсутствующих документов (30 рабочих дней со дня размещения соответствующего уведомления в информационной системе уполномоченного органа (экспертной организации) референтного государства либо со дня направления уведомления заявителю способом, указанным в заявлениях о регистрации и экспертизе) (реш.144, п.25)
3 рабочих дня – УО принимает решение о возврате
заявителю заявлений о регистрации и экспертизе и документов с обоснованием причин возврата, в случае если по истечении 30 рабочих дней заявителем не устранены выявленные нарушения и (или) не представлены отсутствующие документы (реш.144, п.26)
Схема проведения экспертизы регистрационного досье