Основные нормативные правовые акты:
- решение Совета Евразийской экономической комиссии от 12.02.2016 № 27 «Об утверждении Общих требований безопасности и эффективности медицинских изделий, требований к их маркировке и эксплуатационной документации на них» (далее - Общие требования);
- решение Совета Евразийской экономической комиссии от 12.02.2016 № 46 «О Правилах регистрации и экспертизы безопасности, качества и эффективности медицинских изделий» (далее - Правила);
- решение Совета Евразийской экономической комиссии от 10.11.2017 № 106 «О Требованиях к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости от потенциального риска их применения».
Подтверждение безопасности, качества и эффективности медицинского изделия
Перечень документов, необходимых для регистрации медицинского изделия в рамках ЕАЭС и подтверждающих его безопасность, качество и эффективность
- Для всех медицинских изделий
- Документы, доказывающие соответствие медицинского изделия применимым положениям Общих требований
- План сбора и анализа данных по безопасности и эффективности медицинских изделий на постпродажном этапе
- Обязательно для медицинских изделий класса потенциального риска применения 2а (выпускаемых в стерильном виде), 2б или 3
- Отчет об инспекции производства
- Для медицинских изделий, имеющих историю обращения
- Информация о маркетинге, сообщения о несчастных случаях и отзывах
Регистрационное досье. Общие сведения
п. 16 Правил: Для регистрации медицинского изделия заявитель выбирает референтное государство и государства признания
Референтное государство - выбранное заявителем государство-член, уполномоченный орган которого осуществляет регистрацию медицинского изделия
Государство признания - государство-член, уполномоченный орган(экспертная организация) которогоо существляет процедуру согласования экспертного заключения референтного государства. Для процедуры регистрации должно быть выбрано как минимум одно государство признания
Регистрация медицинского изделия осуществляется референтным государством на основании результатов экспертизы медицинского изделия и согласования экспертного заключения государствами признания. (п. 10 Правил)
Заявитель представляет в уполномоченный орган (экспертную организацию) референтного государства следующие документы:
- заявление на проведение экспертизы и регистрации медицинского изделия;
- регистрационное досье, содержащее документы по перечню согласно приложению № 4 к Правилам;
- документы, представленные на иностранном языке, должны иметь заверенный в установленном законодательством государства-члена порядке аутентичный перевод на русский язык;
- все документы регистрационного досье обязательно должны иметь реквизит «дата выдачи документа».
- копии документов, подтверждающих оплату экспертизы и регистрации медицинского изделия в референтном государстве.
В случае если референтным государством является Российская Федерация, то документы принимаются Росздравнадзором по описи. При этом:
- сведения, указанные в описи, должны позволять однозначно соотносить предоставленные заявителем документы с документами, указанными в приложении № 4 к Правилам;
- при представлении в Росздравнадзор документов на бумажном носителе необходимо дополнительно предоставлять сканированные версии данных документов на электронном носителе (в формате.pdf с текстовым слоем с возможностью выделения и копирования блоков, а также осуществления поиска; заявление о проведении экспертизы необходимо представлять как в формате .pdf, так и в формате .doc).
Разделение сканированных версий предоставляемых документов должно проводиться в соответствии с Классификатором видов документов регистрационного досье медицинского изделия, утвержденным решением Коллегии Евразийской экономической комиссии от 03.04.2018 № 48.
Заявление о проведении экспертизы и регистрации медицинского изделия
При оформлении заявлений необходимо учитывать положения следующих нормативных правовых актов:
- Правила
- форма заявлений установлена в приложениях № 2 и 3 к Правилам;
- при одновременной подаче на регистрацию нескольких модификаций медицинского изделия, относящихся к одному виду медицинского изделия в соответствии с применяемой в Союзе номенклатурой медицинских изделий, изготовленных одним производителем, отличающихся друг от друга изменениями комплектации и (или) технических параметров, не влияющими на принцип работы и функциональное назначение, относящихся к одному классу потенциального риска применения, заявитель представляет 1 заявление и 1 регистрационное досье.
В случае если представленные модификации будут относиться к разным видам медицинского изделия в соответствии с указанной номенклатурой, каждая модификация регистрируется отдельно с предоставлением отдельного регистрационного досье. (п. 14 Правил)
- Решение Коллегии Евразийской экономической комиссии от 24.07.2018 № 123 «О Критериях включения в одно регистрационное удостоверение нескольких модификаций медицинского изделия, относящихся к одному виду медицинского изделия в соответствии с применяемой в Евразийском экономическом союзе номенклатурой медицинских изделий»;
- Решение Коллегии Евразийской экономической комиссии от 22.12.2015 № 173 «Об утверждении Правил классификации медицинских изделий в зависимости от потенциального риска применения»;
- Решение Коллегии Евразийской экономической комиссии от 24.07.2018 № 116 «О Критериях разграничения элементов медицинского изделия, являющихся составными частями медицинского изделия, в целях его регистрации»;
- Решение Коллегии Евразийской экономической комиссии от 16.04.2019 № 62 «О классификаторе областей медицинского применения медицинских изделий».
Нюансы при оформлении заявления о проведении экспертизы медицинского изделия
п. 3. Область применения медицинского изделия - заполняется на основании Решения Коллегии ЕЭК от 16.04.2019 № 62.
п. 4. Класс потенциального риска применения медицинского изделия - заполняется на основании Решение Коллегии ЕЭК от 22.12.2015 № 173.
п.5. Код вида медицинского изделия (согласно применяемой в Союзе номенклатуре медицинских изделий) - указывается обязательно, классификатор.
п.7. Комплектация медицинского изделия - заполняется на основании Решений Коллегии ЕЭК от 24.07.2018 № 116 и от 24.07.2018 № 123. Сведения о комплектации должны предоставлять возможность однозначной идентификации модификаций изделия и составных частей.
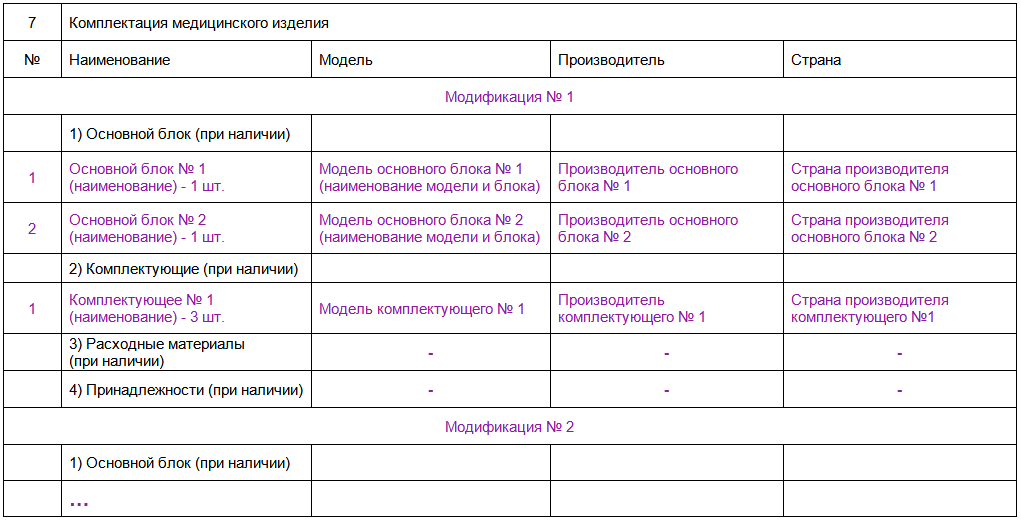
При оформлении заявления о проведении регистрации медицинского изделия
в п. 7 заявления о проведении регистрации медицинского изделия необходимо внести данные из п. 7 заявления о проведении экспертизы медицинского изделия.
п. 10. Регистрация в стране-производителе и других странах - заполняется на основании документов, которые входят в состав досье (пп. 6-7 Приложения № 4 к Правилам).
п. 11. Производство - указываем данные о производстве:
- полностью на данном производстве
- частично на данном производстве
- полностью на другом производстве
п. 12. Сведения о производителе
- в графе "Номер, дата и срок действия разрешительного документа" необходимо указать разрешительный документ, свидетельствующий о регистрации компании в качестве юридического лица (например, для российских компаний - ОГРН и дата его присвоения, ИНН) либо физического лица в качестве индивидуального предпринимателя.
- в графе "Наименование, страна" для иностранных производителей дополнительно рекомендуется указывать сведения о транслитерации наименования.
п. 13. Сведения о производственной(ых) площадке(ах) - в графе "Номер, дата и срок действия разрешительного документа (при наличии)" необходимо указать данные разрешительного документа на право производства.
Производственная площадка - территориально обособленный комплекс, предназначенный для выполнения всего процесса производства медицинского изделия или его определенных стадий.
п. 3 Правил
Заявление о проведении экспертизы медицинского изделия
Под определенными стадиями производственного процесса понимаются:
- изготовление всего медицинского изделия или его основных блоков, кроме основных блоков, являющихся медицинскими изделиями, зарегистрированными в установленном порядке в Союзе, в территориально обособленных комплексах, входящих в организационную структуру производителя медицинского изделия;
- изготовление по договорам со сторонним производителем (подрядчиком) всего медицинского изделия или его основных блоков, кроме основных блоков, являющихся медицинскими изделиями, зарегистрированными в установленном порядке в Союзе.
- стерилизация медицинского изделия.
пп. 1 п. 6 Методических рекомендаций по содержанию и структуре документов регистрационного досье медицинского изделия, утвержденных рекомендацией Коллегии Евразийской экономической комиссии от 08.10.2019 № 2
Заявление о проведении экспертизы медицинского изделия
п. 14. Сведения об уполномоченном представителе (при наличии) - в графе "номер, дата и срок действия разрешительного документа (при наличии)" необходимо указать разрешительный документ, свидетельствующий о регистрации компании в качестве юридического лица (например, для российских компаний - ОГРН и дата его присвоения, ИНН) либо физического лица в качестве индивидуального предпринимателя.
Уполномоченный представитель производителя - юридическое лицо или физическое лицо, зарегистрированное в качестве индивидуального предпринимателя, являющиеся резидентами государства-члена и уполномоченные в соответствии с доверенностью производителя медицинского изделия представлять его интересы и нести ответственность в части обращения медицинского изделия в рамках Союза и исполнения обязательных требований, предъявляемых к медицинским изделиям.
п. 3 Правил
Заявление о проведении экспертизы и регистрации медицинского изделия
В составе досье следует представлять:
- документ, подтверждающий полномочия уполномоченного представителя производителя (в случае наличия данного лица);
- в случае если уполномоченный представитель производителя не является резидентом референтного государства, то рекомендуется представлять копию документа, подтверждающего регистрацию уполномоченного представителя производителя в качестве юридического лица либо индивидуального предпринимателя.
Документы, подтверждающие оплату экспертизы и регистрации медицинского изделия
Размеры государственной пошлины (ст. 333.32.2 Налогового кодекса Российской Федерации).
Российская Федерация – референтное государство
- Выдача регистрационного удостоверения медицинского изделия - 7 000 рублей.
- Проведение экспертизы безопасности, качества и эффективности медицинского изделия (в зависимости от класса потенциального риска его применения в соответствии с правом Союза) при его регистрации:
- класс 1 - 45 000 рублей;
- класс 2а - 65 000 рублей;
- класс 2б - 85 000 рублей;
- класс 3 - 115 000 рублей.
Российская Федерация – государство признания
За согласование экспертного заключения об оценке безопасности, эффективности икачества медицинского изделия при его регистрации (в зависимости от класса потенциального риска его применения в соответствии с Союза):
- класс 1 - 45 000 рублей;
- класс 2а - 65 000 рублей;
- класс 2б - 85 000 рублей;
- класс 3 - 115 000 рублей.
Образцы платежных поручений для оплаты государственной пошлины за совершение действий уполномоченным федеральным органом исполнительной власти при осуществлении регистрации медицинских изделий, предназначенных для обращения на общем рынке медицинских изделий в рамках Союза представлены на официальном сайте Росздравнадзора
Основные замечания
Приложение № 4 к Правилам, п. 9. Данные о маркировке и упаковке (полноцветные макеты упаковок и этикеток, текст маркировки на русском языке и государственных языках государств-членов):
- отсутствие полноцветных макетов упаковок и этикеток;
- представление документа только на русском языке;
- представление информации не в полном объеме;
- несоответствие Общим требованиям.
20 Регистрационное досье Приложение № 4 к Правилам
Приложение № 4 к Правилам, п. 10. Информация о разработке и производстве: схемы процессов производства, основные стадии производства, упаковка, испытания и процедура выпуска конечного продукта
- представление информации не в полном объеме;
- отсутствие указания сведений о производственной(ых) площадке(ах), на которой(ых) осуществляется та или иная часть производственного процесса;
- наличие описания только общих подходов к производству медицинских изделий в целом.
Приложение № 4 к Правилам, п. 11. Сведения о производителе: наименование, вид деятельности, юридический адрес, форма собственности, состав руководства, перечень подразделений и дочерних компаний с указанием их статуса и полномочий
- отсутствие указанных сведений или их представление не в полном объеме;
- представление недостоверных сведений.
В качестве сведений о производителе могут быть представлены копии документа о регистрации юридического лица в соответствии с законодательством страны производителя, устава организации и т.д.
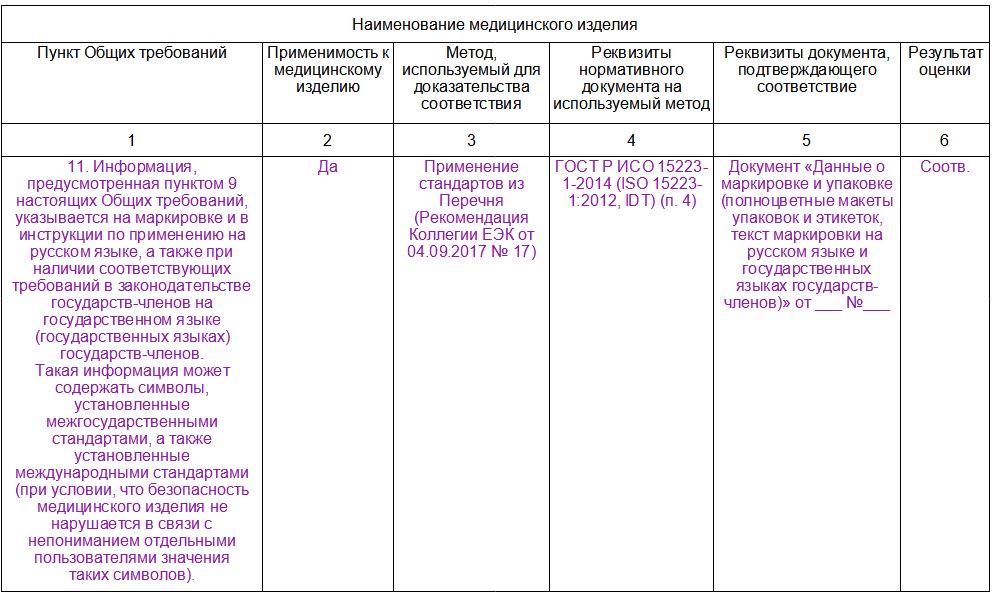
Приложение № 4 к Правилам, п. 15. Сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них.
В целях регистрации соответствие медицинского изделия Общим требованиям подтверждается производителем или его уполномоченным представителем посредством представления в уполномоченный орган государства-члена сведений о соблюдении установленных требований по форме согласно приложению № 2.
п. 111 Общих требований
Порядок заполнения формы представления сведений о соответствии медицинского изделия Общим требованиям
В графе "Применимость к медицинскому изделию":
- для всех МИ в п. 3. - п. 11. Общих требований должен быть утвердительный ответ "да"
- для МИ за исключением МИ для диагностики in vitroв п. 12. - п. 68. Общих требований нужно указать вариант "да" или "нет"
- для МИ для диагностики in vitro п. 69. - п. 108. Общих требований нужно указать вариант "да" или "нет"
В графе "Метод, используемый для доказательства соответствия" необходимо указать:
Доказательства соответствия медицинского изделия положениям, установленным пунктами 3, 6 и 8 Общих требований, должны включать клиническое обоснование на основе клинических данных о медицинском изделии
п. 112 Общих требований
В графе "Реквизиты документа, подтверждающего соответствие": документы, ссылки на которые даны в графе, должны быть представлены в досье.
Пример заполнения формы: