Оценка системы менеджмента качества в рамках Евразийского экономического союза
Документация
- О Требованиях к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости от потенциального риска их применения
Решение совета ЕЭК от 10 ноября 2017 года N 106 - Руководство по оценке и наделению организаций государств – членов Евразийского экономического союза полномочиями по проведению инспектирования производства медицинских изделий
Рекомендация Коллегии Евразийской экономической комиссии от 13 сентября 2021 года № 22 - О требованиях предъявляемых к инспекторам, и порядке установления соответствия инспекторов этим требованиям
Проект решения коллегии ЕЭК от 20.02.2019 года (обсуждение завершено 22.04.2019г.) - О требованиях к инспектирующим организациям, наделяемым полномочиями по проведению инспекций производителей медицинских изделий
Проект решения коллегии ЕЭК от 11.01.2019 года (обсуждение завершено 10.02.2019г.)
Устанавливают в рамках Евразийского экономического союза (далее - Союз) требования к системе менеджмента качества (внедрению, поддержанию) медицинских изделий в зависимости от потенциального риска их применения (раздел II).
Требования к инспектирующим организациям, инспекторам: раздел III, пп. 13 - 19:
- полномочия
- компетенция инспекторов
- конфиденциальность информации
- независимость
Требования к оценке системы менеджмента качества медицинских изделий (раздел III, пп. 20-41).
- система менеджмента качества медицинских изделий - организационная структура, функции, процедуры, процессы и ресурсы, необходимые для скоординированной деятельности по руководству и управлению организацией - производителем медицинских изделий применительно к качеству;
3.5.3 система менеджмента (management system): Совокупность взаимосвязанных или взаимодействующих элементов организации (3.2.1) для разработки политик (3.5.8), целей (3.7.1) и процессов (3.4.1) для достижения этих целей (ГОСТ Р ИСО 9000-2015)
3.3.3 менеджмент (management): Скоординированная деятельность по руководству и управлению организацией (3.2.1) (ГОСТ Р ИСО 9000-2015). - оценка системы менеджмента качества медицинского изделия - подтверждение внедрения, поддержания и результативности функционирования системы менеджмента качества медицинских изделий для обеспечения соответствия выпускаемых в обращение в рамках Союза медицинских изделий применимым к ним Общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке, технической и эксплуатационной документации на них , утвержденным Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 27
- условия производства - инфраструктура и производственная среда, необходимые для обеспечения соответствия производимых медицинских изделий Общим требованиям эффективности и безопасности, требованиям к их маркировке, технической и эксплуатационной документации на них , утвержденным Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 27
- инспектирование производства - оценка условий производства и системы менеджмента качества производителя медицинского изделия на соответствие настоящим Требованиям
- уполномоченный орган - орган государственной власти государства - члена Союза, уполномоченный на осуществление и (или) координацию деятельности в сфере обращения медицинских изделий на территории этого государства
- инспектирующая организация - уполномоченный орган или организация (организации), которой (которым) уполномоченным органом государства - члена Союза делегированы полномочия по проведению инспектирования производства
- критический поставщик - поставщик, продукция или услуги которого оказывают прямое влияние на безопасность и (или) эффективность медицинского изделия
- производственная площадка - территориально обособленный комплекс, предназначенный для выполнения всего процесса производства медицинского изделия или его определенных стадий
- постпродажный мониторинг - система сбора и анализа данных производителя медицинских изделий о применении медицинских изделий, отслеживании и выявлении побочных действий медицинских изделий в процессе их эксплуатации
- недоброкачественное медицинское изделие - медицинское изделие, которое не соответствует общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке, технической и эксплуатационной документации на них и не может быть безопасно использовано по назначению, установленному производителем
п.7 Внедрение СМК, П.4.2.1 ГОСТ ISO 13485-2017
Процессы
- Риски ЖЦП
- Последовательность и взаимосвязь
- Критерии и методы
- Условия производства
- Информационные ресурсы
- Мониторин и измерение
- Анализ
- Меры
п.9 Документация СМК должна содержать в том числе:
- Требования к техническим характеристикам МИ
- Стандарты (методы)
- Учет показателей качества МИ
- Процесс обеспечения качества
- Процесс производства
- Процесс контроля качества
- Процесс корректирующих и предупреждающих действий
- Средства конотроля качества ми и СМК
- Обратная связь с потребителем
- Методы контроля третьей стороны
П.8 Все элементы СМК
должны документально оформляться и поддерживаться в актуальном состоянии.
п.10 СМК необходимо
поддерживать в актуальном состоянии и обеспечивать ее результативность.
п.18 Уполномочивание инспектирующих организаций
на проведение инспекций производителей медицинских изделий осуществляет уполномоченный орган по каждой подгруппе медицинских изделий по перечню в соответствии с приложением N 2 к настоящим Требованиям на основании оценки их соответствия настоящим Требованиям. Уполномочивание инспектирующих организаций на проведение инспекций производителей медицинских изделий осуществляет уполномоченный орган по каждой подгруппе медицинских изделий по перечню в соответствии с приложением N 2 к настоящим Требованиям на основании оценки их соответствия настоящим Требованиям.
- Неактивные МИ, кроме in vitro
- Активные неимплантируемые МИ, кроме in vitro
- Активные имплантируемые МИ
- Медицинские изделия для диагностики in vitro
1. Неактивные МИ (кроме in vitro)
- Сердечно-сосудистые имплантаты
- Ортопедические имплантаты
- Имплантаты мягких тканей
- Функциональные имплантаты
- Зубные имплантаты и стоматологические материалы
- МИ для инъекций, вливания, переливания крови и диализа
- Офтальмологические МИ
- Ортопедические МИ и МИ для реабилитации
- МИ для контрацепции
- едицинские инструменты
- МИ для дезинфекции, гигиенической обработки и стерилизации МИ
- Шовный материал, перевязочные средства и прочие МИ для лечения ран
- МИ не включенные в подгруппы 1.1-1.12
2. Активные неимплантируемые МИ (кроме in vitro)
- МИ для контроля физиологических показателей
- МИ для визуализации с/без ионизирующим излучением
- МИ для лучевой терапии с/без ионизирующим излучением
- МИ для литотрипсии
- МИ для экстракорпорального кровообращения, внутривенного вливания и плазмафереза
- МИ для респираторной терапии
- Наркозно-дыхательные, гипербарические МИ и МИ для стимуляции и ингибирования
- Хирургические МИ
- Офтальмологические МИ
- Стоматологические МИ
- МИ для дезинфекции и стерилизации
- МИ для реабилитации и активные протезы
- МИ для позиционирования и перевозки
- Медицинское самостоятельное ПО
- МИ для экстракорпорального оплодотворения и искусственного оплодотворения
- МИ не включенные в подгруппы
3. Активные имплантируемые МИ
- МИ для стимуляции и ингибирования
- МИ для ввода лекарственных и иных веществ
- МИ поддерживающие, замещающие или заменяющие функции организма
- Радиоактивные имплантаты для внутритканевой лучевой терапии
- МИ не включенные в подгруппы
4. Медицинские изделия для диагностики in vitro
- Реагенты, наборы реагентов, калибровочные и контрольные материалы
- Приборы и оборудование для диагностики in vitro
- Самостоятельное медицинское программное обеспечение для диагностики in vitro
- МИ не включенные в подгруппы
Система менеджмента качества медицинских изделий. Решение совета ЕЭК от 10 ноября 2017 года N 106
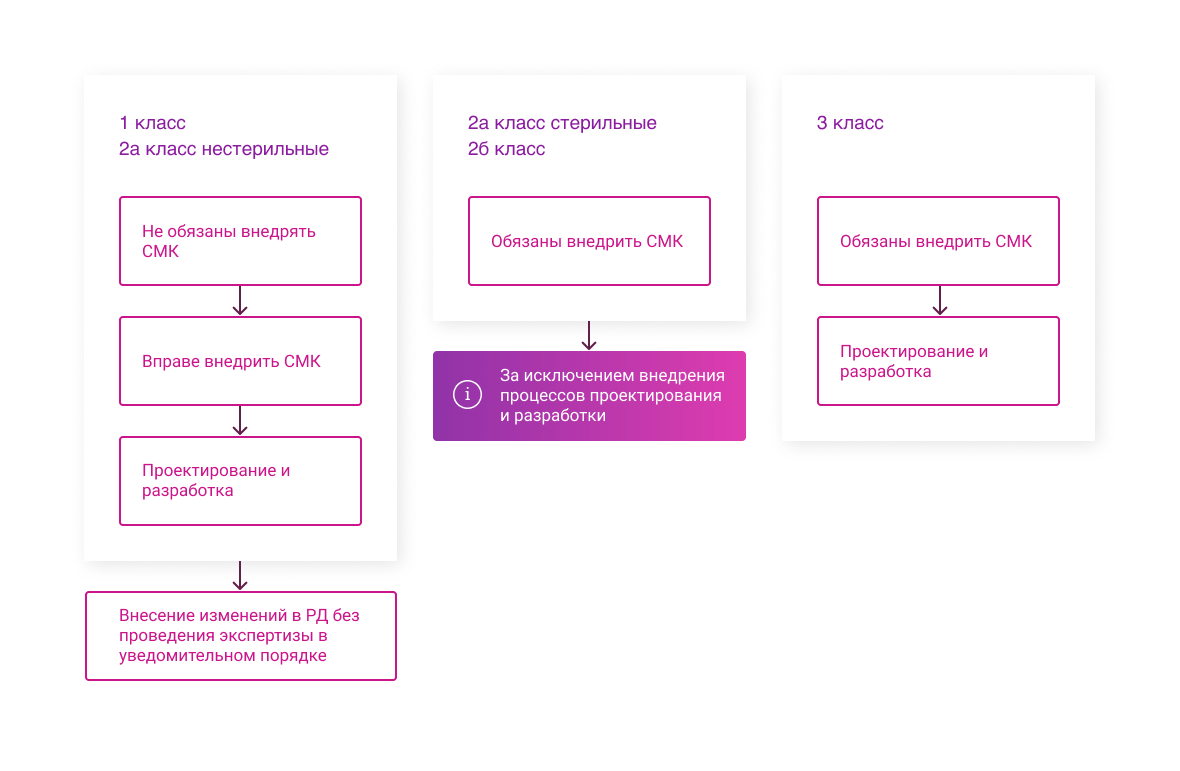
П.18 (продолжение) Уполномоченный орган вправе определять количество инспектирующих организаций для проведения инспектирования производства по заявкам производителей медицинских изделий и (или) в соответствии с графиками проведения инспектирования производства в срок, не превышающий 3 месяца со дня представления производителем медицинского изделия полного комплекта документации, в том числе документов об оплате проведения инспектирования производства.
Уполномоченный орган -> Инспектирующая организация № 1 -> Инспектирующая организация № 2 -> Производство
Формы инспектирования производства
- Первичное при регистрации МИ
- Периодическое (плановое)
- Внеплановое
Процедура инспектирования первичная
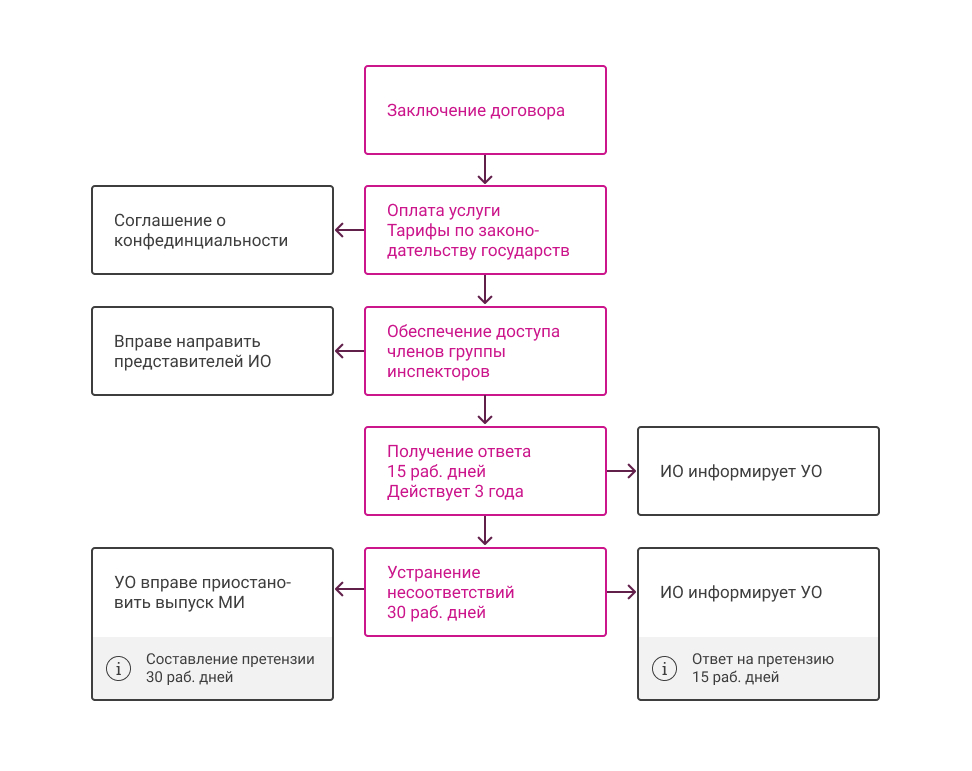
Записи об инспекциях п.17.
17. Инспектирующая организация должна поддерживать в рабочем состоянии и хранить не менее 10 лет записи об инспекциях и другой деятельности по всем производителям медицинских изделий, подавшим заявки о проведении инспектирования производства и (или) прошедшим инспектирование производства, которые должны включать в себя:
- информацию о заявке и отчеты о результатах проведения инспектирования производства
- договор на проведение инспектирования производства
- обоснование продолжительности инспектирования производства
- контроль за выполнением корректирующих действий по результатам проведения инспекций
- записи о жалобах и апелляциях, а также последующих корректирующих действиях
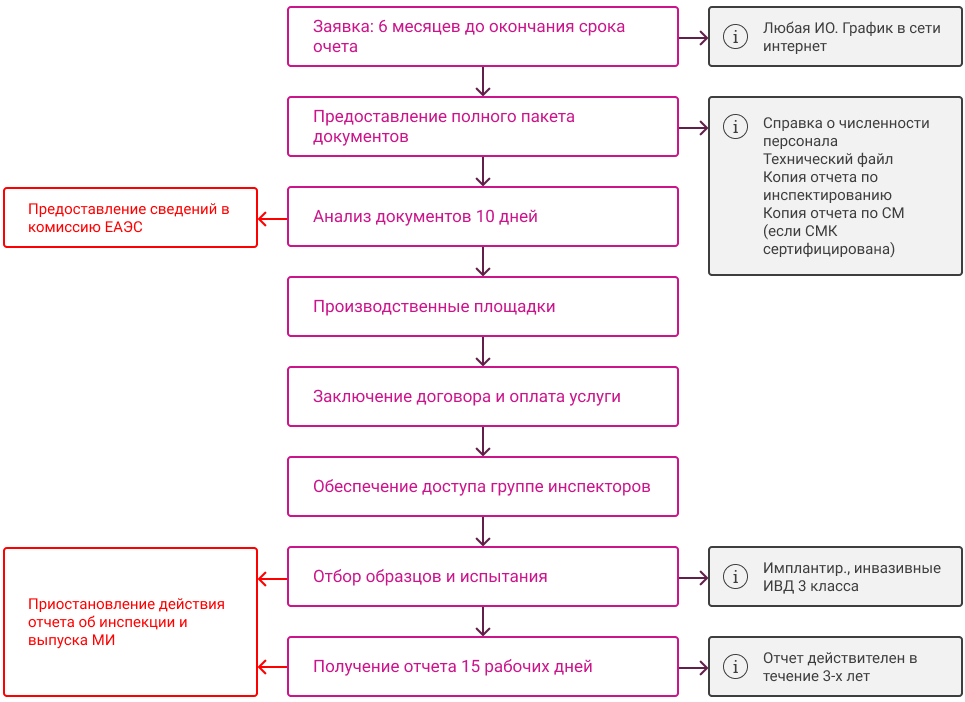
Процедура инспектирования внеплановая
п.40 Производитель вправе обратиться в ИО с заявлением о проведении внепланового инспектирования производства в следующих целях:.
- внесение изменений в перечень групп (подгрупп) МИ
- внесение изменений в перечень производственных площадок
- подтверждение устранения нарушений по результатам проведения инспектирования производства
- подтверждение устранения причин, которые привели к выпуску недоброкачественных медицинских изделий
- подтверждение внедрения производителем системы менеджмента качества медицинских изделий
Результаты инспектирования производства распространяются на группу (подгруппу) медицинских изделий
- Неактивные МИ, кроме in vitro
- Активные неимплантируемые МИ, кроме in vitro
- Активные имплантируемые МИ
- Медицинские изделия для диагностики in vitro
Оценка системы менеджмента качества МИ
п.24. При проведении инспектирования производства проводится оценка системы менеджмента качества медицинских изделий для следующих процессов:
- Управление документацией и записями
- Проектирование и разработка, если есть в СМК
- Производство
- Выходной контроль
- Корректирующие и предупреждающие средства
- Связь с потребителем
Проектирование и разработка
- подтверждение наличия процедур проектирования и разработки (включая управление рисками)
- анализ документов, описывающих процедуру проектирования и охватывающих модельный ряд медицинского изделия
- подтверждение на основе выбранных записей по проектированию медицинского изделия того, что процедуры проектирования и разработки были установлены и применены
- подтверждение того, что деятельность по менеджменту риска была определена и осуществлена, критерии допустимости риска были установлены и являются соответствующими, любой остаточный риск был оценен и при необходимости доведен до сведения потребителя
Управление документацией и записями
- подтверждение того, что процедуры идентификации, хранения и уничтожения (включая управление изменениями) были разработаны
- подтверждение наличия документов, необходимых для того, чтобы организация могла обеспечивать планирование, осуществление производственных процессов и управление ими
- подтверждение того, что документация на МИ включает в себя:
- свидетельства соответствия МИ установленным требованиям
- описание МИ, включая инструкции по применению, материалы и спецификацию
- сводную документацию по верификации и валидации проектов (в том числе данные клинических исследований (испытаний)
- маркировку медицинских изделий
- документы по менеджменту риска
Производство
- анализ производственных процессов изготовления серийной продукции (включая условия производства)
- оценку процессов стерилизации (для медицинских изделий, выпускаемых в стерильном виде), в том числе:
- определение того, что процессы стерилизации были документированы, записи параметров процесса стерилизации для каждой стерилизуемой партии медицинских изделий поддерживаются в рабочем состоянии
- определение того, что процесс стерилизации был валидирован
- определение того, что процесс стерилизации проводится в соответствии с установленными параметрами
- подтверждение того, что процессы производства являются управляемыми и контролируемыми и функционируют в установленных пределах, а также подтверждение обеспечения необходимого уровня контроля продукции и (или) услуг критических поставщиков
- подтверждение идентификации и прослеживаемости медицинских изделий и процессов их производства, а также их соответствия установленным требованиям
- подтверждение того, что деятельность по выходному контролю медицинских изделий обеспечивает соответствие медицинских изделий установленным требованиям и была документирована.
Выходной контроль
Корректирующие и предупреждающие действия
- подтверждение того, что процедуры корректирующих и предупреждающих действий были разработаны
- подтверждение того, что средства управления препятствуют распространению медицинских изделий, качество которых не соответствует Общим требованиям безопасности и эффективности медицинских изделий
- подтверждение того, что корректирующие и предупреждающие действия являются результативными
- подтверждение того, что производитель медицинского изделия разработал эффективную процедуру выпуска и применения уведомлений по безопасности медицинских изделий
Связь с потребителем
- подтверждение того, что производитель МИ принял меры, необходимые для установления связи с потребителями, в целях выполнения необходимых корректирующих и предупреждающих действий, располагает системой сбора и анализа данных о безопасности и эффективности МИ на постпродажном этапе и поддерживает ее в актуальном состоянии, а также направляет в уполномоченный орган отчеты о результатах постпродажного мониторинга безопасности и эффективности МИ в соответствии с Правилами проведения мониторинга безопасности, качества и эффективности МИ
- подтверждение того, что обратная связь с потребителем анализируется производителем МИ в ходе процессов жизненного цикла продукции и используется для повторной оценки риска и при необходимости для актуализации деятельности по менеджменту риска
п.37. При проведении периодического (планового) инспектирования производства оцениваются
- поддержание соответствия системы менеджмента качества медицинского изделия настоящим Требованиям
- результативность системы менеджмента качества медицинских изделий в обеспечении соответствия выпускаемых в обращение в рамках Союза медицинских изделий применимым к ним Общим требованиям безопасности и эффективности
ГОСТ ISO 13485-2017
- Наличие Политики и целей в области качества
- Наличие Руководства по качеству
- В части управления документацией (правила выпуска и пересмотра)
- Наличие процедуры проведения анализ со стороны руководства
- Наличие Представителя со стороны руководства
- Выполнение требований потребителя
- Наличие процедуру по управлению человеческими ресурсами (ответственность, полномочия, компетентность)
- Валидация процессов производства (кроме процесса стерилизации)
- Наличие процедур по управлению собственности потребителя
- Наличие процедур по улучшению
- Наличие процедуры проведения внутреннего аудита СМК
| Фактическая численность сотрудников (человек) | Нормативная продолжительность первичного инспектирования производства (человеко-дней) | Нормативная продолжительность периодического (планового) инспектирования производства (человеко-дней) |
|---|---|---|
| 5-49 | 6 | 4 |
| 50-99 | 7 | 5 |
| 100-199 | 8 | 6 |
| 200-499 | 9 | 7 |
| 500-999 | 10 | 8 |
| 1000-1999 | 11 | 9 |
| 2000-4999 | 12 | 10 |
| более 5000 | 13 | 11 |
Классификация несоответствий
-
I этап – предварительная оценка применение классификационной матрицы
-
II этап – окончательная оценка применение повышающих баллов
I этап оценки - Классификационная матрица
- прямое влияние, если касается требований, относящихся к процессам проектирования, разработки, производства и выходного контроля медицинского изделия.
- непрямое влияние, если затрагивает требования, связанные с функционированием системы менеджмента качества медицинских изделий.
| Влияние несоответствия на безопасность, эффективность и качество медицинского изделия |
прямое | 3 | 4 |
| непрямое | 1 | 2 | |
| впервые | повторно | ||
| Повторяемость несоответствия | |||
впервые означает, что конкретное несоответствие не было выявлено в ходе 2 последних случаев проведения инспектирования производства,
повторно означает, что конкретное несоответствие было выявлено в ходе одного из 2 последних случаев проведения инспектирования производства
II этап – окончательная оценка применение повышающих баллов
- Остутствие документированных процедур, относящихся к процессам проектирования, разработки, производства и выходного контроля медицинского изделия, а также к постпродажному мониторингу, необходимых для обеспечения безопасности и эффективности медицинского изделия
- Выпуск в обращение недоброкачественного медицинского изделия в течение отчетного периода. В случае если производителем медицинского изделия проведено внеплановое инспектирование производства в целях подтверждения устранения причин, которые привели к выпуску недоброкачественного изделия, повышающий балл не начисляется
Интегральная оценка несоответствия
| N п/п |
Несоответствие | Градация несоответствия | |||
| I этап, балл | II этап при отсутствии ДП, балл | II этап при выпуске недоброкачест- венного МИ, балл |
суммарный балл | ||
| 1 | 4 | да + 1 балл | нет | 5 | |
Инспектирующая организация не должна выносить положительное заключение о соблюдении производителем медицинского изделия настоящих Требований, если одно или более нарушений оценены на 5 или 6 баллов или более двух нарушений оценены на 4 балла.
Если у Вас остались вопросы по инспектированию производства обратитесь к нам
Подробнее о нашей услуге по внедрению СМК и подготовке производства к инспектированию можно ознакомиться здесь


